 |
| Prototipo "Baby Blue" de termociclador del año 1986. Fuente. |
Pero por si existiera aún alguna duda sobre la existencia del VIH, existe una herramienta aún más poderosa si cabe: la reacción en cadena de la polimerasa o PCR. Esta herramienta, cuya función es conseguir una cantidad de ADN suficiente como para poder trabajar con él a partir de una muestra con un contenido muy bajo en ADN puede utilizarse para amplificar fragmentos concretos de un ADN muy concreto. En este caso, se puede amplificar el genoma del VIH. Y sí, sé que el VIH es un virus ARN, es algo que ya he comentado. Pero la PCR ya no es la misma que en 1986 y ha mejorado hasta el punto de poder incluso cuantificar el ARN de la muestra inicial de la que partíamos al principio.
PCR: describiendo la técnica
La técnica de la PCR tiene su origen en 1971, cuando el grupo de Gobind Khorana descubrió una manera de copiar cortas secuencias de ADN usando una polimerasa aislada para obtener muchas copias de estos oligonucleótidos. Sin embargo, fue Kary Mullis, en 1984, quien, mientras intentaba desarrollar un nuevo método para detectar mutaciones, acabó dándose cuenta de que había descubierto una forma de conseguir numerosas copias de fragmentos de ADN utilizando secuencias cebadoras o primers aprovechándose de las polimerasas que copian el ADN.
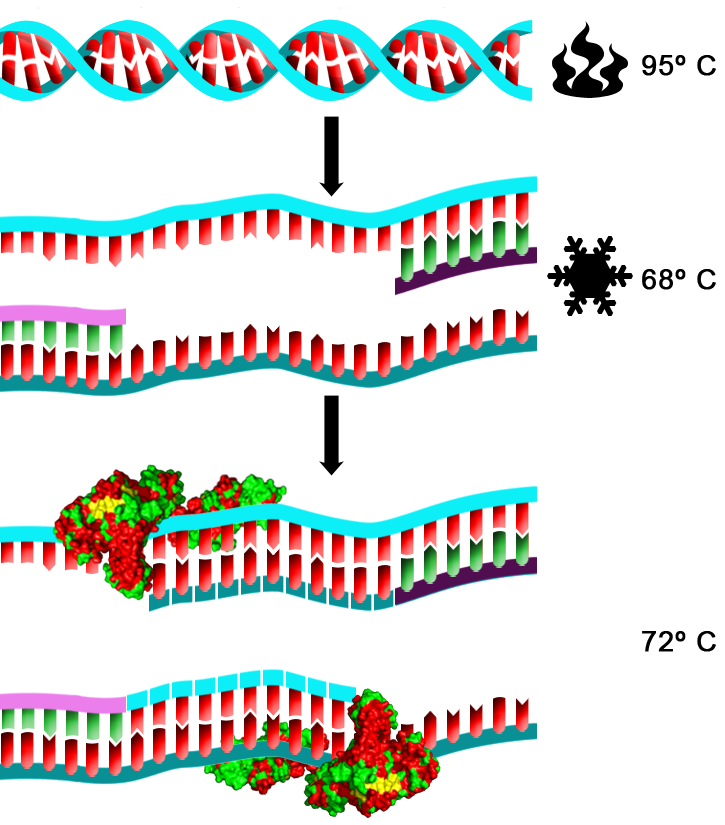 |
| Esquema del primer ciclo de un experimento de PCR |
Es hora de enfriar la mezcla, pero no del todo, para evitar una renaturalización completa. La temperatura de reacción bajará hasta los 68º C. Esto va a permitir que los fragmentos más grandes de ADN no se unan entre sí, pero sí los más pequeños. De esta manera, los cebadores que comentábamos antes se van a unir a las secuencias a las que son complementarios. Esto es, si el cebador tiene una secuencia 3'-AATGCCGT-5' se va a unir a una secuencia 3'-ACGGATT-5'.
Esta será la señal para que la ADN polimerasa actúe. Pero existe un problema. Si bajamos la temperatura hasta los 37º C, que es la temperatura a la que trabajan la mayoría de las polimerasas, el ADN va a renaturalizar, expulsando del amplicón, que es como se llama al fragmento de ADN amplificado por PCR, a los cebadores y formándose de nuevo la molécula de ADN original, con lo que no habríamos amplificado nada. Es por ello que utilizamos ADN polimerasas especiales que funcionan a altas temperaturas y que son resistentes a la desnaturalización o pérdida de estructura tridimensional por calor. La que se utilizó originalmente, y aún se utiliza por su facilidad de acceso y su bajo coste, es la ADN polimerasa de una bacteria termófila, Thermus aquaticus. A esta ADN polimerasa se la denomina Taq polimerasa. Su temperatura óptima de trabajo está en torno a los 70º C. Es por ello que la temperatura, que estaba a 68º C se eleva ligeramente hasta los 72º C. Esto permite dos cosas. La primera, que la Taq polimerasa trabaje a gusto. Y la segunda, que si se han formado bucles intracatenarios o dupletes inespecíficos entre las hebras de ADN separadas y/o los cebadores, se deshagan, quedando fijados únicamente los cebadores que se unieron a sus secuencias diana. La polimerasa entrará en esos lugares y copiará la cadena de ADN complementaria junto al cebador, originándose así dos cadenas de ADN iguales a la original.
 |
| Resultado de un experimento de PCR tras n ciclos de amplificación. |
Mejoras de la técnica
Cuando Kary Mullis describió la técnica por primera vez, utilizó, como hemos apuntado antes, una ADN polimerasa de un organismo que es capaz de vivir a altas temperaturas. Pero la Taq polimerasa tiene una tasa de error comparable a la ADN polimerasa de Escherichia coli, de alrededor de 1 fallo cada 106 nucleótidos copiados. Para fragmentos cortos es posible que esta tasa de error no sea significativa, pero cuando se trata de secuenciar un genoma o determinar la presencia de un patógeno se requiere una fidelidad mucho más alta. Actualmente, existen polimerasas, bien clonadas a partir de sus organismos de origen, bien fusionando dos proteínas distintas mediante ingeniería genética, que mejoran la tasa de error de la Taq polimerasa. Por ejemplo, la Pfu polimerasa, que proviene de la bacteria Pyrococcus furiosus, o la Pwo polimerasa, que proviene de Pyrococcus woesei, tienen tasas de error que son 10 veces mejores que las de la Taq polimerasa. Entre las polimerasas de fusión encontramos las llamadas Phusion High Fidelity Polimerases que pueden llegar, en su tercera generación, a mejorar hasta 52 veces esa tasa de error.
Además de la mejora en la fidelidad de la copia, la técnica actual es mucho más rápida. La automatización del proceso, mediante la utilización de termocicladores ha reducido el tiempo de experimentación y obtención de muestras significativamente. Un ejemplo: hace 15 años, el proceso de PCR de un gen de secalina del centeno (Secale cereale), duraba toda una noche y requería un aceite especial que permitiera la transmisión correcta del calor desde el aparato al tubo de reacción. Actualmente, el proceso dura dos horas y media y se hace en seco.
Pero las mejoras más significativas han venido de la mano de la RT-PCR y la qPCR. La primera, la RT-PCR parte de ARNm para conseguir cadenas de ADN bicatenario mediante la aplicación de una retrotranscriptasa de un virus de aves o de ratones. Esto permite amplificar y extraer ADN a partir de cualquier tránscrito que se esté produciendo en una célula en un momento dado. La qPCR emplea fluorescencia unida a los cebadores, de forma que, al comparar la reacción de amplificación con una reacción control cuantificada, se puede determinar el número de copias de una molécula de ADN concreta que había en la muestra original. Incluso se han llegado a combinar ambas técnicas para generar una técnica denominada RT-qPCR, cuyo resultado no es sino el número de moléculas de ARNm que había en la muestra original.
Además de la mejora en la fidelidad de la copia, la técnica actual es mucho más rápida. La automatización del proceso, mediante la utilización de termocicladores ha reducido el tiempo de experimentación y obtención de muestras significativamente. Un ejemplo: hace 15 años, el proceso de PCR de un gen de secalina del centeno (Secale cereale), duraba toda una noche y requería un aceite especial que permitiera la transmisión correcta del calor desde el aparato al tubo de reacción. Actualmente, el proceso dura dos horas y media y se hace en seco.
Pero las mejoras más significativas han venido de la mano de la RT-PCR y la qPCR. La primera, la RT-PCR parte de ARNm para conseguir cadenas de ADN bicatenario mediante la aplicación de una retrotranscriptasa de un virus de aves o de ratones. Esto permite amplificar y extraer ADN a partir de cualquier tránscrito que se esté produciendo en una célula en un momento dado. La qPCR emplea fluorescencia unida a los cebadores, de forma que, al comparar la reacción de amplificación con una reacción control cuantificada, se puede determinar el número de copias de una molécula de ADN concreta que había en la muestra original. Incluso se han llegado a combinar ambas técnicas para generar una técnica denominada RT-qPCR, cuyo resultado no es sino el número de moléculas de ARNm que había en la muestra original.
El diagnóstico de VIH por PCR
Como habéis podido comprobar, la PCR es una técnica muy útil para obtener grandes cantidades de ADN a partir de ADN o ARNm. Para experimentación supone una herramienta muy potente. Pero, ¿podría aplicarse al diagnóstico?
Volvamos a nuestro ciclo de PCR, el que os representaba más arriba. Fijaos bien en el segundo paso, en el que enfriamos la reacción a 68º C. ¿Qué ocurre en este paso? En la mezcla de reacción hay unos cebadores que se unirán a la cadena original de ADN por complementariedad. Y la unión será tanto mejor cuanto más complementarias sean las secuencias del ADN a amplificar y el cebador.
 |
| Los cebadores de PCR son complementarios a secuencias presentes en el ADN a amplificar |
Si tenemos en cuenta esto, podemos diseñar cebadores de ADN que sean complementarios a secuencias que nos interesen. Pensadlo. Si encontramos un cebador con capacidad de unirse única y exclusivamente a una secuencia determinada, lo que la Taq polimerasa amplifique partiendo de él será única y exclusivamente la secuencia que nos interesa. Pues en esto mismo es en lo que se basa el test de detección de VIH por PCR.

El protocolo de detección del VIH mediante PCR es de tipo RT-PCR, esto es, incluye la retrotranscripción del ARN del virus. Así, a partir de una muestra de sangre del paciente, se extraen linfocitos T del paciente que se han dividido hasta tener un número de células que sea suficiente como para obtener ARN del virus. De este cultivo, se obtiene ARN total.
Usando retrotranscriptasa del virus de la leucemia murina de Moloney, lo transformamos en ADN.

Será este ADN el que amplificaremos usando cebadores específicos de los tres genes env, gag y pol. En este test se utilizan cebadores para tres genes específicos del VIH: env, gag y pol, que corresponden a proteínas de la envuelta, la cápsida y la transcriptasa inversa del virus. Estos genes no tienen ningún homólogo en el ser humano con el que puedan confundirse, por lo que cualquier fragmento de ADN que amplifiquemos usando los cebadores específicos de esos tres genes va a ser un fragmento de ADN que proviene del virus. Ninguno de los cebadores de un gen va a unirse al gen que no le corresponda, por lo que en la misma muestra se pueden incluir los tres pares de cebadores a la vez para amplificar los tres genes a la vez.
 |
| Gel resultado del test VIH. En los carriles en los que están presentes las tres bandas es en los que se puede diagnosticar sin lugar a dudas la presencia del VIH. |
¿Recordáis que habíamos puesto únicamente cebadores para genes del virus y que estos son los únicos que se iban a amplificar? Pues siendo así, las moléculas fluorescentes con las que habíamos marcado el gel, se van a unir a estas moléculas de ADN, acumulándose, por lo tanto, en la banda de peso molecular de los genes del VIH. En un paciente con VIH es precisamente eso lo que obtenemos: un gel en el que se encuentran bandas de tamaños moleculares correspondientes a los genes env, gag y pol.
Pero cabe la posibilidad de que alguno de los cebadores encuentre una secuencia complementaria y amplifique algo que no debe. En este caso, la banda ocupará en el gel un peso molecular que no le corresponde, por lo que no podría tomarse como una marca para el diagnóstico. Pero vayamos más allá y pensemos que es verdad, que podemos amplificar algo con un cebador que no corresponde y con un tamaño molecular similar a lo que estamos buscando. El hecho de que pasemos el test con 3 genes que sólo están en el VIH, nos sirve para discriminar que las muestras en las que está el VIH son sólo aquellas en las que tengamos las tres bandas de amplificación, y no una o dos.
Es decir, que si utilizamos los cebadores específicos de genes que son únicos del VIH, lo que vamos a amplificar son genes específicos del VIH, con lo que podemos obtener un diagnóstico que, unido a ELISA y Western Blot, confirman la presencia del VIH en un paciente.
hola...! pregunto por que nuestro exentrico protagonista y creador de la tecnica PCR promulga que su maquina no sirve para detectar VIH y siendo un "premio novel" es un disidente de la version oficialista...?
ResponderEliminarNo, agustin, Kary Mullis no dice que su "máquina" (él lo que describió fue la técnica, no la máquina) no sirva para detectar el VIH. Sino que la técnica no debería aplicarse de la forma que se aplica, que su aplicación es errónea. Los resultados han demostrado que él se equivocaba.
Eliminarno entiendo como le dieron el premio novel a alguien que se equivoque en algo por lo que justamente le dieron ese premio... Me gustaria tener mas informacion al respecto,tu tienes, la podrias postear.
ResponderEliminarNo, Mullis no se equivocó en la técnica. Se equivocó en las apreciaciones que hizo. Tal como pongo en este artículo, él buscaba una forma de secuenciar rápida y se encontró con una forma de obtener grandes cantidades de ácidos nucleicos a partir de muestras reducidas. Para él, esta es la única aplicación de su técnica que es válida. No concibe que alguien haya podido idear algo que a él se le ha escapado.
EliminarEsto lo contaba en una entrevista hace tiempo. Buscaré a ver si la encuentro.
Hola buenas interesante blog,una pregunta.
ResponderEliminarPorque dicen los expertos que la técnica de PCR no es buena para diagnosticar infecciones y sin embargo es la que usan los bancos de sangre??
La combinan con otras pruebas??
Un saludo
La PCR se usa en combinación con otras pruebas. Su valor diagnóstico depende de la especificidad de los cebadores que se usen. Un ejemplo: para VIH los cebadores son muy específicos. Pero para diagnosticar infecciones por bacterias u otros virus, con una diversidad mucho mayor, se necesitan otras pruebas que confirmen qué cepa de microbio tenemos entre manos.
Eliminar